251. (7.13) Исполнитель должен понимать, что работы, передаваемые на аутсорсинг, включая проведение анализа по договору, проверяются уполномоченными федеральными органами исполнительной власти.
Договор
252. (7.14) Между заказчиком и исполнителем должен быть заключен договор, в котором рекомендуется определить их взаимные обязательства и процедуры передачи информации, связанные с деятельностью, передаваемой на аутсорсинг. Технические аспекты договора должны составляться лицами, имеющими соответствующие знания, связанные с деятельностью, передаваемой на аутсорсинг, и настоящими Правилами. Согласованные сторонами условия договора и все связанные с договором технические и иные соглашения, должны соответствовать законодательству Российской Федерации и регистрационному досье.
253. (7.15) В договоре рекомендуется указывать, какая из сторон отвечает за каждый этап деятельности, передаваемой на аутсорсинг (например, управление знаниями, перенос технологии, обеспечение цепи поставок, заключение договора с третьей стороной, закупку исходного сырья, материалов и их качество, проведение испытаний и выдачу разрешения на использование исходного сырья и упаковочных материалов, проведение производства и контроля качества (включая контроль в процессе производства, отбор образцов и их анализ).
254. (7.16) Все записи, связанные с деятельностью, передаваемой на аутсорсинг, например, записи производства, анализа и реализации продукции, а также соответствующие контрольные образцы должны храниться у заказчика, или заказчик должен иметь к ним доступ. Любые записи, относящиеся к оценке качества продукции, в случае предъявления претензий, предполагаемого несоответствия требованиям или при расследовании в случае предположения о фальсификации продукции должны быть доступны заказчику и точно определены в его соответствующих процедурах.
255. (7.17) В договоре рекомендуется предусматривать право заказчика на аудит передаваемой на аутсорсинг деятельности, которая выполняется исполнителем или взаимно согласованной третьей стороной.
Претензии и отзыв продукции (Глава 8)
Принцип
256. Все претензии и информация, касающиеся потенциально недоброкачественных лекарственных средств, тщательно расследуются в соответствии с утвержденными процедурами. Производитель имеет систему быстрого и эффективного отзыва с рынка продукции с выявленными или предполагаемыми нарушениями качества.
Претензии
257. (8.1) Производитель должен назначить работника, ответственного за рассмотрение претензий и принятие решений, имеющего право привлекать необходимый персонал. Если этот работник не является уполномоченным лицом, то уполномоченное лицо должно быть поставлено в известность обо всех фактах предъявления претензий, расследований и отзывов продукции.
258. (8.2) Действия по рассмотрению претензий на потенциально недоброкачественные лекарственные средства и по принятию решения об отзыве продукции должны быть изложены в соответствующих утвержденных процедурах.
259. (8.3) Любая претензия по качеству продукции должна быть зарегистрирована с указанием исходных данных и тщательно расследована. К этой работе, как правило, следует привлекать лицо, ответственное за контроль качества продукции.
260. (8.4) Если обнаружено или подозревается несоответствие качества какой-либо серии продукции установленным требованиям, необходимо принять решение о проверке аналогичных серий, а также тех серий, которые могут включать продукты, полученные при переработке недоброкачественной серии.
261. (8.5) Решения и меры, принятые по результатам рассмотрения любой претензии, должны быть зарегистрированы и включены в соответствующее досье на серию.
262. (8.6) Записи рассмотрения претензий должны регулярно анализироваться с целью выявления специфических и повторяющихся факторов, которые требуют особого внимания и могут привести к отзыву продукции.
263. (8.7) Особое внимание необходимо уделять оценке того, является ли причиной претензии фальсификация продукции.
264. (8.8) В случае если производитель предпринимает действия, являющиеся следствием возможных ошибок в производстве, ухудшения качества продукции, выявления фальсифицированной продукции или других серьезных проблем, связанных с качеством продукции, должен быть проинформирован соответствующий уполномоченный федеральный орган исполнительной власти.
Отзыв продукции
265. (8.9) Производитель должен назначить работника, ответственного за своевременный отзыв продукции с рынка, имеющего право привлекать необходимый персонал. Как правило, этот работник должен быть независимым от подразделений реализации и маркетинга. Если этот работник не является уполномоченным лицом, то уполномоченное лицо должно быть осведомлено обо всех фактах отзыва продукции.
266. (8.10) Порядок отзыва продукции должен быть регламентирован утвержденной процедурой, которую следует регулярно проверять и при необходимости пересматривать.
267. (8.11) Отзыв продукции должен осуществляться оперативно и в любое время.
268. (8.12) Компетентные органы всех стран, куда была направлена продукция, должны быть немедленно информированы о принятии решения об отзыве продукции в связи с подозрением или обнаружением несоответствия ее качества.
269. (8.13) Записи по отгрузке должны быть доступны лицу (лицам), ответственному(ым) за отзыв продукции, и содержать достаточную информацию об организациях оптовой торговли лекарственными средствами и прямых заказчиках (адреса, номера телефонов и (или) факсов в рабочее и в нерабочее время, номера серий и объемы поставок), включая экспортные поставки и поставки образцов лекарственных средств.
270. (8.14) Отозванную продукцию необходимо промаркировать и хранить отдельно в безопасной зоне вплоть до принятия решения о ее дальнейшем использовании или уничтожении.
271. (8.15) Последовательность действий при отзыве продукции должна быть оформлена документально. Окончательный отчет должен содержать материальный баланс между количеством поставленной и возвращенной продукции.
272. (8.16) Эффективность мероприятий по отзыву продукции должна регулярно анализироваться.
Самоинспекция (Глава 9)
Принцип
273. Самоинспекция проводится с целью проверки выполнения производителем требований настоящих Правил и предложения необходимых корректирующих действий.
274 (9.1) Вопросы, касающиеся персонала, помещений, оборудования, документации, технологического процесса, контроля качества, реализации лекарственных средств, мероприятий по работе с претензиями, отзывов продукции, а также деятельности по проведению самоинспекций, должны регулярно анализироваться в соответствии с заранее утвержденной программой по определенному графику для проверки их соответствия принципам фармацевтической системы качества.
275. (9.2) Самоинспекция должна проводиться независимо и тщательно специально назначенными квалифицированнымми лицами, состоящими в штате производителя. При необходимости может быть проведен независимый аудит производителя экспертами сторонних организаций.
276. (9.3) Результаты самоинспекций должны быть оформлены документально. Отчеты, составленные по результатам самоинспекций, должны включать в себя всю полученную информацию и необходимые корректирующие действия (где применимо). Действия, предпринимаемые по результатам проведенных самоинспекций, также следует оформлять документально.
IV. Основные требования к фармацевтическим субстанциям, используемым в качестве исходного сырья (Часть II)
Введение (1)
277. Юридические лица, на имя которых выданы регистрационные удостоверения, и производители должны использовать в качестве исходного сырья только те фармацевтические субстанции, которые произведены с соблюдением настоящих Правил.
Цель (1.1)
278. Настоящая глава устанавливает требования, касающиеся надлежащего производства фармацевтических субстанций с соответствующей системой управления качеством. Она также предназначена для целей обеспечения качества и чистоты действующих веществ в соответствии с предъявляемыми к ним требованиями.
279. В настоящей главе термин "производство" включает в себя все виды операций с фармацевтическими субстанциями: приемку материалов, производство, упаковку, переупаковку, маркировку, перемаркировку, контроль качества, выдачу разрешения на выпуск, хранение и реализацию, а также соответствующие меры контроля. Требования, установленные настоящей главой, являются обязательными, если Приложениями к настоящим Правилам не установлены иные требования, подлежащие применению в соответствующих случаях, а также если соблюдение указанных требований не может быть заменено альтернативными действиями, обеспечивающими по крайней мере, эквивалентный уровень качества продукции.
280. Настоящие Правила не устанавливают требования, предъявляемые при регистрации фармацевтических субстанций. Производитель обязан выполнять все требования, установленные при включении фармацевтических субстанций в государственный реестр лекарственных средств.
Область применения (1.2)
281. Настоящая глава устанавливает требования к производству фармацевтических субстанций, используемых в производстве лекарственных препаратов для медицинского и ветеринарного применения, в том числе к производству фармацевтических субстанций, получаемых с использованием донорской крови или плазмы в качестве исходного сырья. Она применяется ко всем исходным действующим веществам совместно с Приложениями NN 2 - 7 к настоящим Правилам, где содержатся дополнительные требования для определенных видов действующих веществ.
282. Пункты 622 - 644 настоящих Правил содержат требования, которые распространяются только на производство фармацевтических субстанций, используемых для получения лекарственных препаратов, предназначенных для клинических исследований.
283. К производству стерильных фармацевтических субстанций требования настоящей главы применимы только до стадии стерилизации.
284. Настоящая глава не распространяется на процессы стерилизации и производства стерильных фармацевтических субстанций в асептических условиях. Такие процессы должны проводиться в соответствии с принципами настоящих Правил, требованиями, изложенными в Приложении N 1 к настоящим Правилам, а также в иных нормативных правовых актах Российской Федерации.
285. Настоящая глава не распространяется на цельную донорскую кровь и плазму, поскольку требования по взятию и испытанию крови определяются соответствующими нормативными правовыми актами Российской Федерации, а также на нерасфасованные лекарственные препараты ("ангро", "ин балк"). Если нормативными правовыми актами Российской Федерации установлены специальные требования для обеспечения качества при производстве лекарственных средств для ветеринарного применения против эктопаразитов, такие требования должны соблюдаться.
286. Исходное сырье для производства фармацевтических субстанций - это исходное сырье, промежуточные продукты или другие фармацевтические субстанции, которые используют в производстве фармацевтических субстанций и которые как важный структурный фрагмент вводят в структуру фармацевтической субстанции. Исходное сырье для производства фармацевтических субстанций может приобретаться по договору у одного или нескольких поставщиков либо производиться самостоятельно. Исходное сырье для производства фармацевтических субстанций, как правило, имеет установленные химические свойства и структуру.
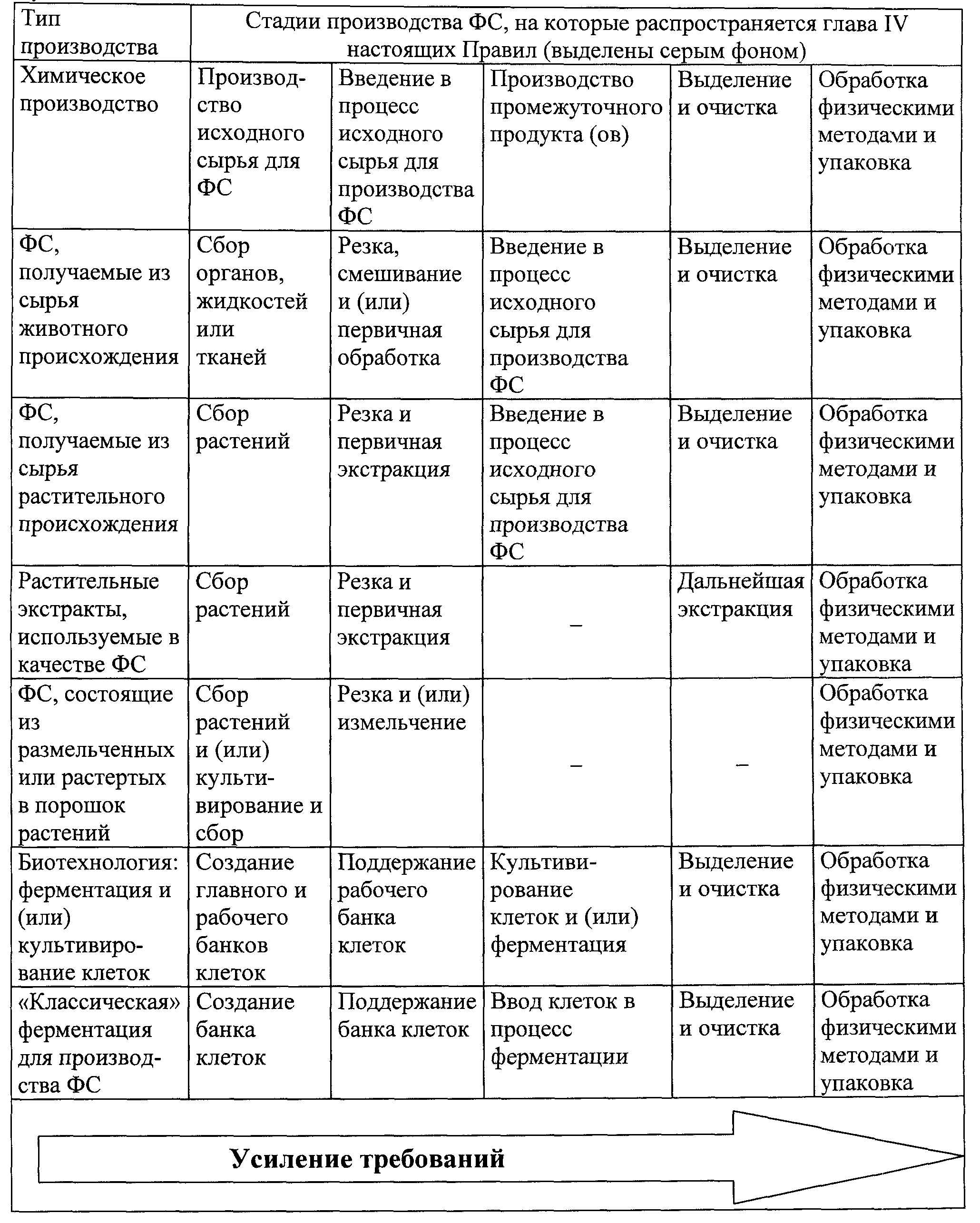
287. Производитель должен определить и документально обосновать стадию, с которой должно начинаться производство фармацевтической субстанции. Для процессов синтеза эта стадия определяется как стадия ввода в технологический процесс исходного сырья для производства фармацевтических субстанций. Для других процессов (например, для ферментации, экстракции, очистки) данную стадию определяют с учетом конкретных особенностей производства. В таблице N 1 приведены требования относительно момента, когда обычно вводят в процесс исходное сырье для производства фармацевтических субстанций. Начиная с этой стадии, на данные промежуточные продукты и (или) стадии производства фармацевтических субстанций распространяются требования настоящей главы. Они включают в себя валидацию критических стадий производственного процесса, оказывающих влияние на качество фармацевтических субстанций. В то же время выбор производителем стадии технологического процесса для проведения валидации не обязательно означает, что эта стадия является критической. Требования настоящей главы распространяются, как правило, на стадии, выделенные в таблице N 1 серым фоном. Это не означает, что в процессе производства должны выполняться все стадии, указанные в данной таблице. Строгость следования требованиям настоящей главы должна возрастать от ранних стадий производства фармацевтических субстанций к завершающим стадиям технологического процесса, очистки и упаковки. Обработку физическими методами фармацевтических субстанций, такую как грануляция, покрытие оболочкой или физическое изменение размера частиц (например, грубый и тонкий помол), следует проводить, по крайней мере, в соответствии с требованиями настоящих Правил. Настоящая глава не применяется к стадиям, которые предшествуют введению в процесс веществ, определенных как исходное сырье для производства фармацевтических субстанций.
288. В настоящей главе используется термин "фармацевтическая субстанция" (ФС), который следует рассматривать как взаимозаменяемый с термином "активный фармацевтический ингредиент" (АФИ). Термины, употребляемые в настоящей главе, и определения обозначенных ими понятий (которые приведены в пункте 645 настоящих Правил) используются только для целей настоящей главы.
 | |
| 2060 × 2571 пикс. Открыть в новом окне | |